
Overall research program. Our research program is focused on the molecular machines and intricate mechanisms responsible for genome replication, genome maintenance and gene expression. Computational biology has emerged as a powerful tool for understanding these complex systems, significant for human health. To define inherently dynamic biological processes, my group combines computational methods with data from structural biology techniques that can probe flexible macromolecular systems – cryo-electron microscopy (cryo-EM), small angle X-ray scattering (SAXS) and single-molecule Förster Resonance Energy Transfer (sm-FRET). Known high-resolution structures of constituents in these complexes are integrated with cryo-EM, SAXS, sm-FRET and biochemical data to yield information on the larger assemblies through hybrid computational protocols we are developing. In this context, we are ideally positioned to take advantage of the exciting recent advances in cryo-EM that have boosted resolution to near-atomic level. In collaboration with EM groups, we have applied novel computational techniques to model structures (e.g. transcription initiation complexes in various functional states) using 3 – 6 Å resolution cryo-EM maps. We also collaborate with structural biologists and single-molecule spectroscopy experts to uncover the dynamics of key elements of the DNA replication machinery. Such close interplay of computation and experiment is needed for in-depth mechanistic and functional analyses.
Specific areas of research.
 Understanding the inner workings of the replisome – the complex molecular machine that accomplishes replication of chromosomal DNA – is undeniably among the great challenges in the biomedical sciences and could impact fundamental knowledge of the causes of cancer, degenerative neurological and inherited genetic disorders. Ongoing work in this area involves new strategies to model constituents of the replisome (sliding clamp proteins and their assemblies with core replication proteins and cognate DNA). The project leverages hybrid computational methods and the experience of established experimental collaborators. Moving forward, we aim to tackle larger assemblies in DNA replication initiation and potentially model entire replisomes from cryo-EM data. Particularly exciting have been the recent in vitro reconstitution of a functional eukaryotic replisome (comprised of 31 individual proteins) and the groundbreaking cryo-EM determination of a complete bacteriophage replisome. Thus, deciphering the molecular organization of complete replisomes, including eukaryotic ones, has come within reach. We are well positioned to capitalize on these advances and contribute to the breakthroughs that are likely to shape the replication field in the next decade.
Understanding the inner workings of the replisome – the complex molecular machine that accomplishes replication of chromosomal DNA – is undeniably among the great challenges in the biomedical sciences and could impact fundamental knowledge of the causes of cancer, degenerative neurological and inherited genetic disorders. Ongoing work in this area involves new strategies to model constituents of the replisome (sliding clamp proteins and their assemblies with core replication proteins and cognate DNA). The project leverages hybrid computational methods and the experience of established experimental collaborators. Moving forward, we aim to tackle larger assemblies in DNA replication initiation and potentially model entire replisomes from cryo-EM data. Particularly exciting have been the recent in vitro reconstitution of a functional eukaryotic replisome (comprised of 31 individual proteins) and the groundbreaking cryo-EM determination of a complete bacteriophage replisome. Thus, deciphering the molecular organization of complete replisomes, including eukaryotic ones, has come within reach. We are well positioned to capitalize on these advances and contribute to the breakthroughs that are likely to shape the replication field in the next decade.
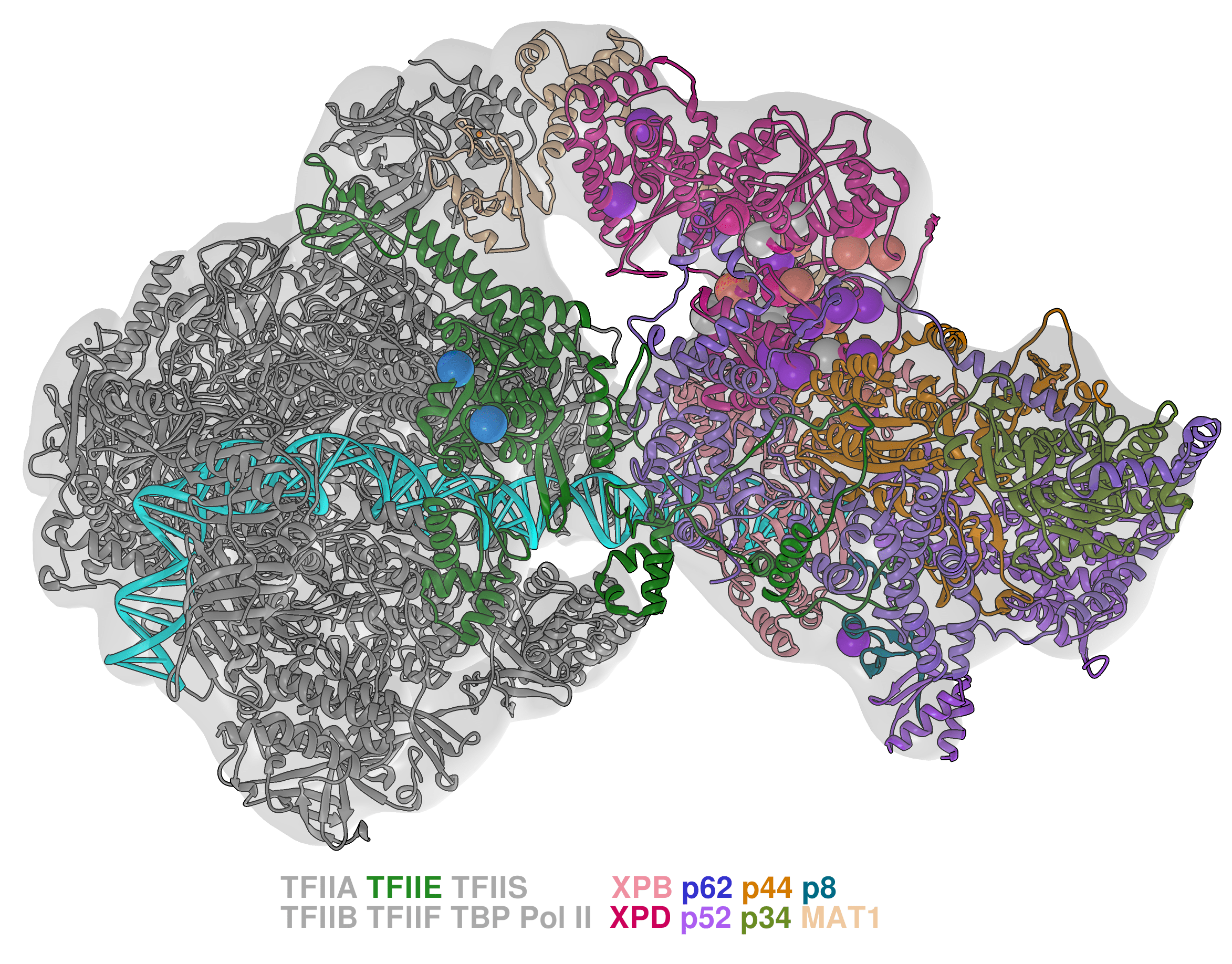 Modeling molecular machines in gene regulation. RNA Polymerases (Pol I, II and III) constitute the centerpieces of the intricate molecular machinery that transcribes the genetic code into RNA and controls such diverse processes as cell differentiation, development and responses to environmental change. My group aims to provide new structural and mechanistic understanding of these machineries relying on parallel advances in cryo-EM and computational modeling. Just as computational methods are providing exciting advances for protein design and fold prediction, they also have the potential to powerfully link structures to functional dynamics and biological phenotypes. I believe it is timely to rethink approaches to cryo-EM structural analysis and how to better incorporate disease mutations into structural models and elucidate essential dynamic communities within functional complexes. We will work to 1) determine how the Pol I, II and III transcription machineries recognize and open promoter DNA; 2) examine the role in transcription initiation of the TFIID transcription factor, which serves as a platform to assemble the transcription preinitiation complex (PIC); 3) delineate the function of the transcription complexes in controlling gene expression. Our aim is to uncover the mechanisms and allosteric networks that transmit regulatory signals to the RNA Polymerases. Success of this project will have major biomedical impacts – both in understanding disease etiology and in providing a structural framework to devise effective treatments.
Modeling molecular machines in gene regulation. RNA Polymerases (Pol I, II and III) constitute the centerpieces of the intricate molecular machinery that transcribes the genetic code into RNA and controls such diverse processes as cell differentiation, development and responses to environmental change. My group aims to provide new structural and mechanistic understanding of these machineries relying on parallel advances in cryo-EM and computational modeling. Just as computational methods are providing exciting advances for protein design and fold prediction, they also have the potential to powerfully link structures to functional dynamics and biological phenotypes. I believe it is timely to rethink approaches to cryo-EM structural analysis and how to better incorporate disease mutations into structural models and elucidate essential dynamic communities within functional complexes. We will work to 1) determine how the Pol I, II and III transcription machineries recognize and open promoter DNA; 2) examine the role in transcription initiation of the TFIID transcription factor, which serves as a platform to assemble the transcription preinitiation complex (PIC); 3) delineate the function of the transcription complexes in controlling gene expression. Our aim is to uncover the mechanisms and allosteric networks that transmit regulatory signals to the RNA Polymerases. Success of this project will have major biomedical impacts – both in understanding disease etiology and in providing a structural framework to devise effective treatments.
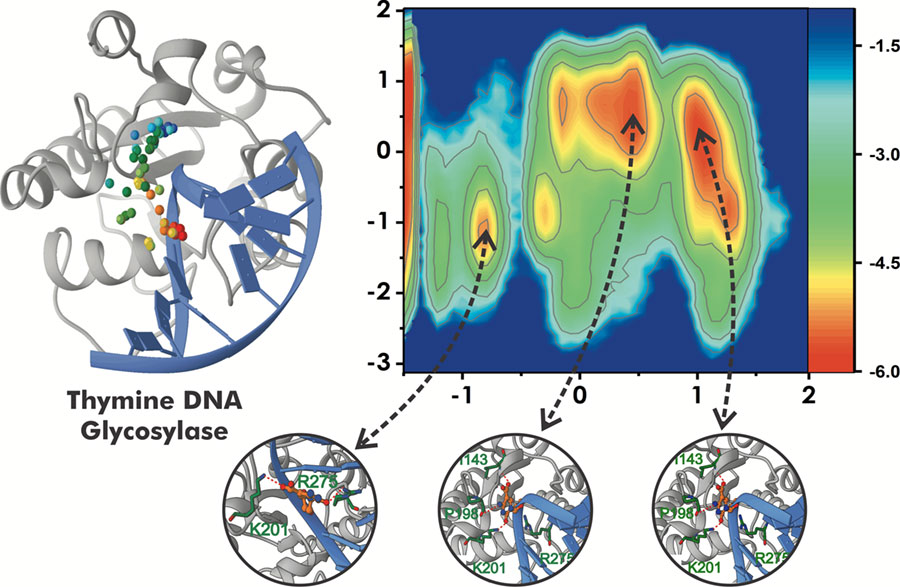
Modeling pathways and enzymes involved in epigenetic regulation. We also seek to expand our footprint in the area of epigenetic regulation beyond current efforts on the mechanisms and inhibition of protein arginine methyltransferase enzymes. To this end, we have established a new line of research that will leverage path optimization and enhanced sampling methods to elucidate the complex interplay of enzymes with dual roles in DNA repair and epigenetic regulation (DNA and RNA methylation/demethylation pathways). Genome maintenance occurs in the context of chromatin and it is becoming increasingly apparent that epigenetic regulation is intricately intertwined with the DNA damage response. Understanding how epigenetic marks are recognized, distinguished from exogenous or endogenous DNA lesions, and processed by the canonical DNA repair machinery is a topic of great current interest. We will devise computational strategies to uncover the mechanisms of concerted action of DNA glycosylases and repair nucleases in these pathways while benefiting from synergistic experimental efforts with established collaborators.
In summary, advanced computational methods will be combined with insightful experimental collaborative efforts to understand the function of replication and transcription complexes as vital components of molecular machines engaged in safeguarding genome stability.
Research highlights

A highlight of our work on DNA replication from the Oak Ridge Leadership Computing Facility.

A highlight of our work on DNA repair and epigenetic regulation from the San Diego Supercomputer Center.
A highlight of our work on transcription initiation from the Texas Advanced Computing Center.
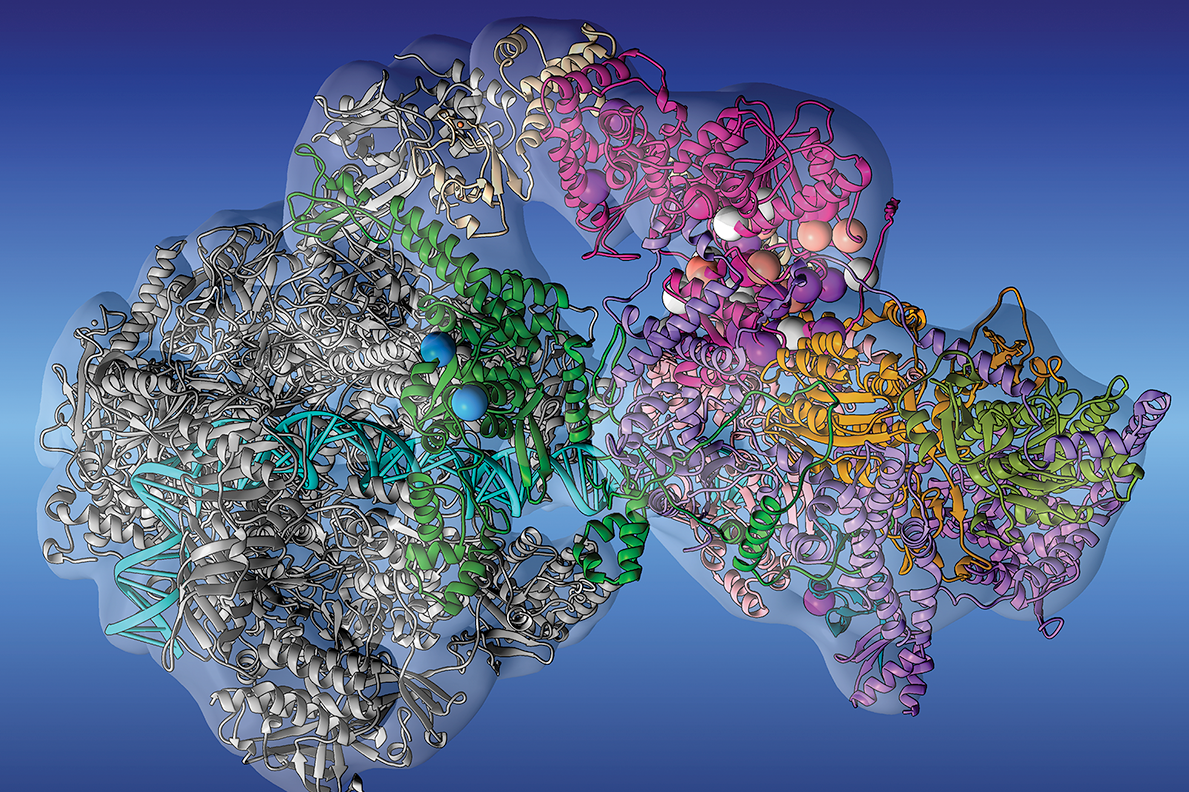
A highlight of our work on the origins of genetic diseases from the Oak Ridge Leadership Computing Facility.
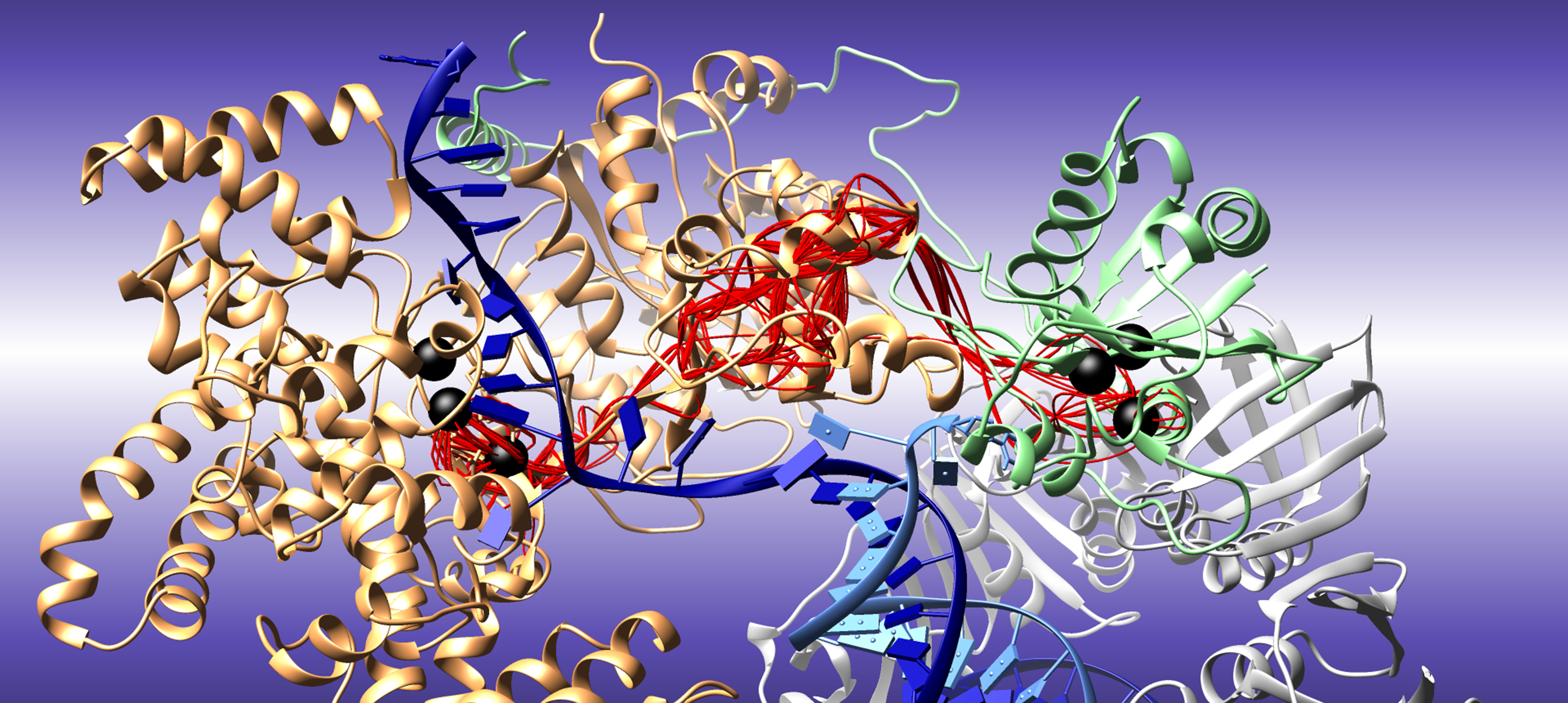
A highlight of our work on high-fidelity DNA replication from the Oak Ridge Leadership Computing Facility.
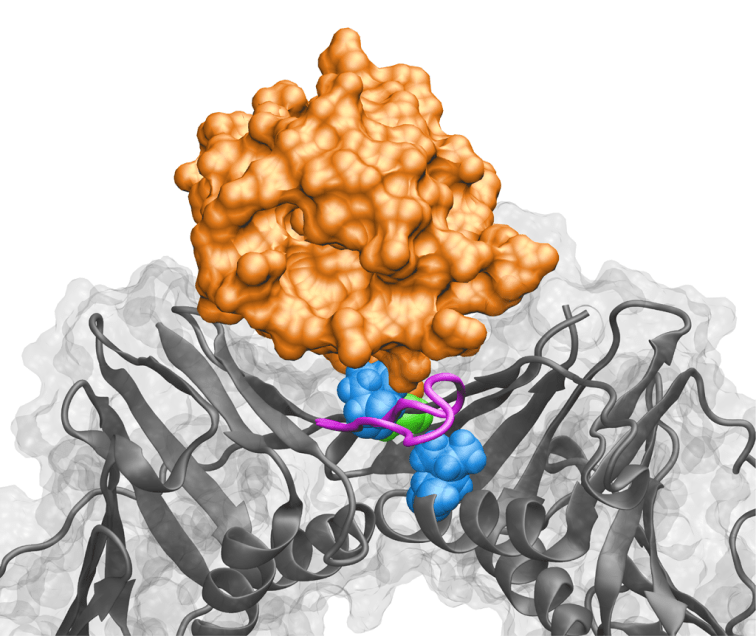
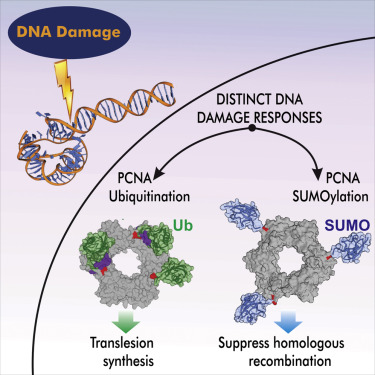
A highlight of our work on posttranslational modifications in the DNA damage response from the Oak Ridge Leadership Computing Facility.
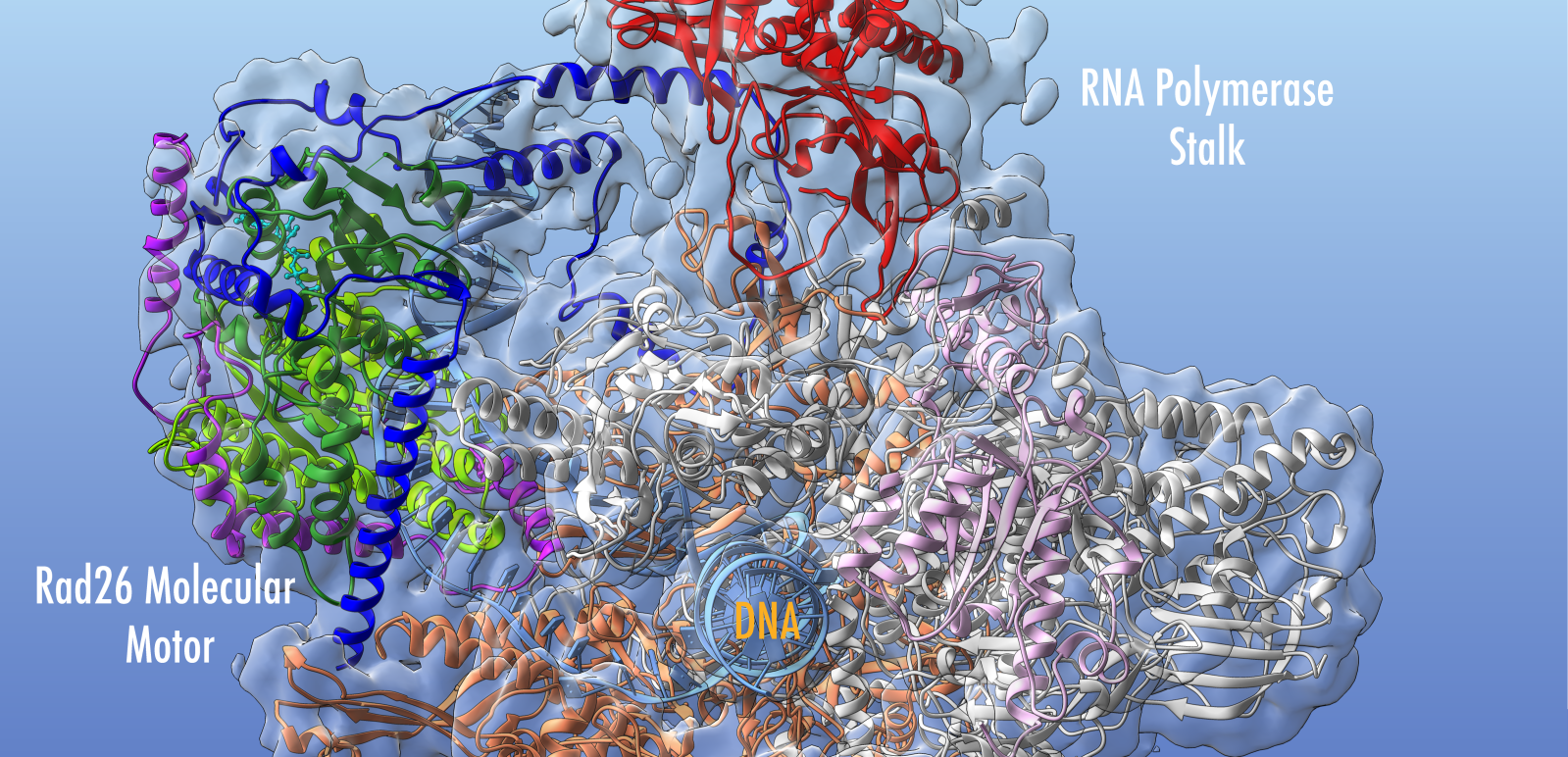
A highlight of our work on transcription-coupled nucleotide excision repair from the Oak Ridge Leadership Computing Facility.
